
E-mail: info@tlph.com.la
Among over 7,000 known rare diseases, pulmonary arterial hypertension (PAH) stands out as a paradox: affecting more than 25 million people globally, it is an "uncommon" condition with startling prevalence. Primarily caused by genetic mutations or heart-related disorders, PAH leads to abnormally high blood pressure in the pulmonary arteries, forcing the body into chronic oxygen deprivation. For patients, even the simple act of breathing becomes a relentless fight for survival.
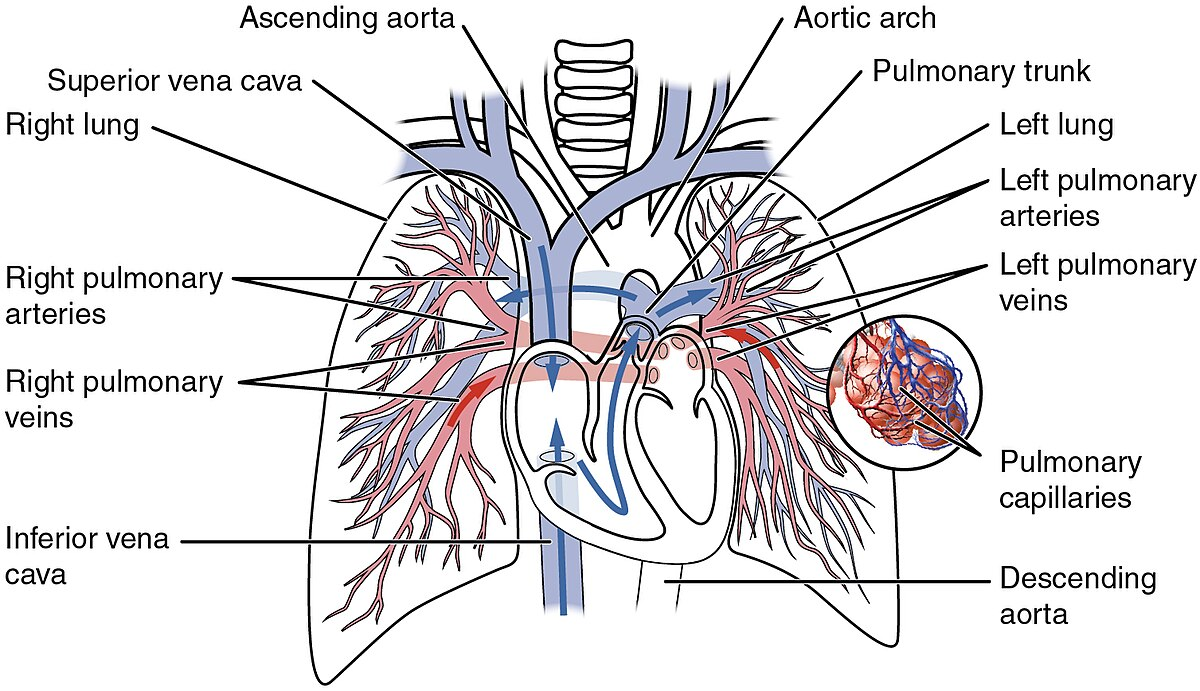
Classified by the World Health Organization (WHO) into five subtypes, pulmonary hypertension (PH) includes:
Arterial PAH (vascular origin),
PH due to left heart disease,
PH linked to lung diseases or hypoxia,
Chronic thromboembolic PH,
PH with unclear or multifactorial causes.
PAH’s insidious onset and symptom overlap often lead to misdiagnosis, while its high mortality rate and risk of severe complications—such as heart failure—pose grave threats. Despite its prevalence, awareness remains critically low. Highlighting PAH is not just about medical urgency; it’s a call to recognize the invisible battles millions face daily. By amplifying understanding and advocacy, we transform “rarity” into visibility, ensuring no breath—and no life—is overlooked.
Macietan and Tadalafei produced by Tongmeng(Lao)(TLPH) have been approved for market by Lao FDD!
Macitentan: As a dual endothelin receptor antagonist (ERA), it inhibits the binding of endothelin-1 (ET-1) to both ETA and ETB receptors, thereby suppressing ET-1-mediated vasoconstriction, smooth muscle proliferation, and fibrosis. This reduces pulmonary vascular resistance and slows disease progression.
Tadalafil: A phosphodiesterase-5 (PDE5) inhibitor, it increases cyclic guanosine monophosphate (cGMP) levels by inhibiting PDE5 enzyme activity, enhancing nitric oxide (NO)-mediated vasodilation and improving pulmonary hemodynamics.
Impact and Therapeutic Urgency of PAH
Pulmonary arterial hypertension (PAH) is a rare, progressive, and life-threatening disease characterized by narrowing of pulmonary arterioles, elevated pulmonary arterial pressure, and eventual right heart failure. Symptoms such as dyspnea and fatigue are nonspecific, often leading to delayed diagnosis (typically 2–4 years), by which time the disease is advanced. PAH pathogenesis involves dysregulation of multiple pathways, including endothelin, NO-cGMP, and prostacyclin. Current guidelines recommend early combination therapy targeting these pathways to delay progression. However, complex regimens with multiple daily pills challenge adherence, underscoring the need for simplified therapies like fixed-dose combinations .








 Address: Rd.13 South, 31 km Ban Naphasuk,
SaithanyDistrict, Vientiane, Lao PDR
Address: Rd.13 South, 31 km Ban Naphasuk,
SaithanyDistrict, Vientiane, Lao PDR  E-mail: info@tlph.com.la
E-mail: info@tlph.com.la  WeChat: TLPH01 Whatsapp:+856 20 76 814 389
WeChat: TLPH01 Whatsapp:+856 20 76 814 389  Website: https: //tlph.com.la
Website: https: //tlph.com.la